Currently, most single cell protocol require the preparation of a single cell suspension from fresh tissue, a major roadblock to clinical deployment, to archived materials and to certain tissues such as adult brain. In the adult brain the harsh enzymatic dissociation harms the integrity of the cells and their RNA, and biases toward easily dissociated cell types, and is restricted to young animals.
Our DroNc-Seq method is based on the Drop-Seq method (Macosko et al. Cell 2015), which is the first massively parallel single cell RNA-seq method. We combined this sNuc-Seq (Habib et al. 2016) for analysis of single nuclei from fresh, frozen or lightly fixed tissues. sNuc-Seq offers three critical benefits: (1) handling complex tissues and cells that are very challenging to dissociate; (2) handling minute samples; (2) access to archived samples, and ability to collect a sample in one location and process it in another. DroNc-seq, a hybrid of the above two methods, combines the strengths of the nuclei approach with the strength of droplet microfluidics, which enables profiling thousands of cells at very low cost.
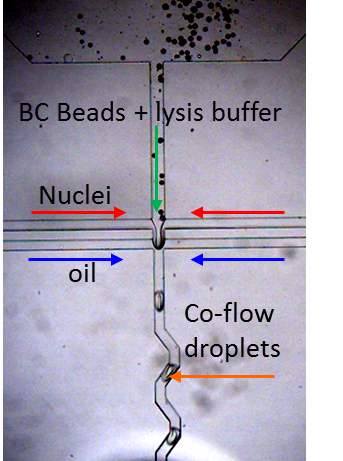
To perform single nuclei RNA-seq in droplets we modified the microfluidic device design with commensurate flow parameters to accommodate the small size of the nuclei, nuclei isolation protocol and downstream PCR conditions. We generate ~75 um drops at 3 kHz speed to co-encapsulate single nuclei with Drop-Seq barcode beads for in-drop nuclear lysis and mRNA capture onto BC beads, followed by droplet breakage and RT in bulk, a la Drop-Seq. A smaller droplet volume translates to a higher concentration of mRNA in these drops (more than 5x) compared to those used in Drop-Seq. However, the overall design and workflow are maintained such that it can be easily performed by anyone currently working with Drop-Seq with minimal adjustments to the protocol.
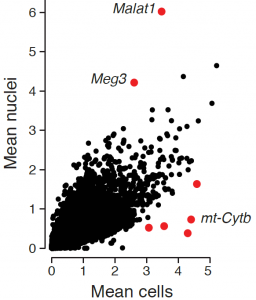
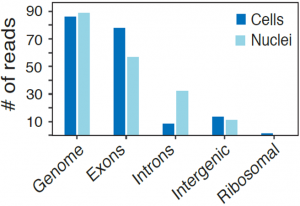
While the average expression profile of single nuclei was highly correlated with the average profile of single cells (Pearson r=0.87, figure above, left), genes with significantly higher expression in nuclei (e.g., the lncRNAs Malat1 and Meg3) or cells (mitochondrial genes Mt-nd1, Mt-nd2, Mt-nd4, Mt-cytb) were consistent with their known distinct enrichment in nuclear vs. non-nuclear compartments. Interestingly, while in both methods over 85% of reads align to coding loci, in cells 80% of these reads map to exons, while in nuclei 56% map to exons and 32% to introns (figure above, right), reflecting the enrichment of nascent, pre-processed transcripts in the nuclear compartment.
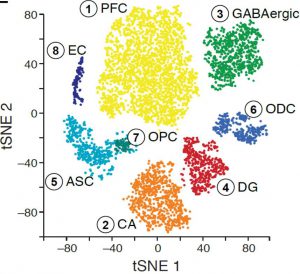
We applied DroNc-Seq to the adult mouse Hippocampus and Prefrontal Cortex brain regions (frozen tissue). Analysis of DroNc-Seq data revealed distinct nuclei clusters corresponding to known cell types and anatomical distinctions between the brain regions and within the hippocampus (figure, left). Moreover, we could distinct between the different types of glia cells, including Astrocytes, Oligodendrocytes and Oligodendrocytes Precursor Cells despite their relatively low level of RNA and the resulting low level of genes detected (figure left). With DroNc-seq we maintain the ability to detect cell types and marker genes, while increasing the throughput.
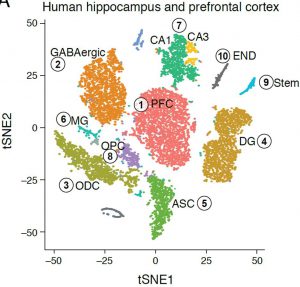 Similar analysis of the adult human hippocampus and prefrontal cortex brain regions (frozen archived sample), revealed distinct nuclei clusters corresponding to known cell types and anatomical distinctions (figure, right). We were also able to capture finer distinctions between closely related cells, such as distinguishing between sub-types of GABAergic neurons, demonstrating the sensitivity and strength of the DroNc-Seq method. Notably, the human archived samples vary in quality, but with DroNc-Seq we were able to get high-quality libraries of both neurons and glia cells from each sample, demonstrating the robustness of our method and its utility for clinical applications.
Similar analysis of the adult human hippocampus and prefrontal cortex brain regions (frozen archived sample), revealed distinct nuclei clusters corresponding to known cell types and anatomical distinctions (figure, right). We were also able to capture finer distinctions between closely related cells, such as distinguishing between sub-types of GABAergic neurons, demonstrating the sensitivity and strength of the DroNc-Seq method. Notably, the human archived samples vary in quality, but with DroNc-Seq we were able to get high-quality libraries of both neurons and glia cells from each sample, demonstrating the robustness of our method and its utility for clinical applications.

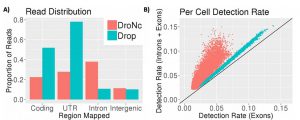
Since its publication, we have incorporated intronic reads into our alignment and pre-processing pipeline. We find that up to 50% of the reads from DroNc-seq map to intronic regions, while for Drop-seq, only 7% of reads were intronic (figure below, A). Including these intronic reads to quantify gene expression level improves the gene detection rate in DroNc-seq by ~2 times on average (figure below, B). This increase in detection rate leads to recovery of gene expression for cells which would otherwise not be detected and can be used to compensate for the smaller amount of nuclear RNA compared with a similar Drop-seq run on whole cells.